St. Jude Family of Websites
Explore our cutting edge research, world-class patient care, career opportunities and more.
St. Jude Children's Research Hospital Home

- Fundraising
St. Jude Family of Websites
Explore our cutting edge research, world-class patient care, career opportunities and more.
St. Jude Children's Research Hospital Home
- Fundraising
Taosheng Chen Lab
Decoding the transcriptional regulation of therapeutic responses
About the Chen Lab
The body’s defense system is finely tuned to detect, destroy, or export endogenous or foreign toxic substances. While this system has obvious protective benefits, it can also contribute to treatment failure by decreasing drug efficacy and increasing drug resistance and toxicity. Our laboratory uses promiscuous xenobiotic nuclear receptors as a model to investigate the transcriptional regulation of response to therapeutics. Using small molecules to modulate these receptors, we are able to control their role in drug metabolism to help improve clinical intervention.

Our research summary
Using hypothesis-driven and technology-enabled approaches, our laboratory develops biology methods and chemical probes to investigate the chemical regulation of transcription, the molecular basis of signal crosstalk among transcription factors and the structural basis of selective modulation of highly homologous enzymes. Notably, we use the promiscuous nuclear receptors pregnane X receptor (PXR) and constitutive androstane receptor (CAR) as models. PXR and CAR transcriptionally regulate cytochrome P450 3A4 (CYP3A4) and CYP3A5 —enzymes that metabolize more than 50% of clinical drugs. PXR and CAR are promiscuously activated by structurally diverse compounds. While activation of the PXR/CAR-CYP3A4/CYP3A5 pathway protects the body against toxic compounds, it decreases drug efficacy and induces drug toxicity and resistance. Our central goal is to understand the transcriptional and enzymatic mechanisms governing therapeutic responses and design strategies to modulate them to overcome adverse drug responses.
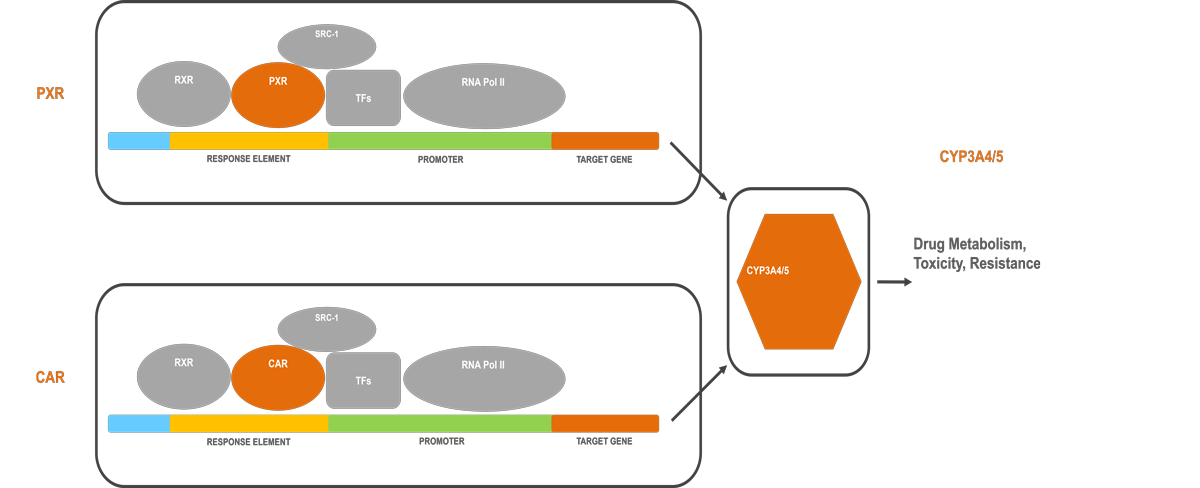
Our work is not without its hurdles. The ligand promiscuity and structural flexibility of PXR and CAR have hampered the development of their selective chemical modulators, the lack of which has been a critical barrier to investigating their transcriptional regulation; the absence of a defined mechanism to explain the contradiction between the perception (that PXR and CAR are functionally redundant) and the observations (that they are mutually inhibitory) presents a challenge; and the high structural similarity between CYP3A4 and CYP3A5 and their promiscuous ligand binding pockets have prevented the development of selective modulators.

To tackle these challenging problems, our laboratory integrates model systems and multidisciplinary experimental approaches, including cell, molecular, biochemical, biophysical, pharmacological, structural and chemical strategies and high-throughput technologies. We initiate by developing bioassays that enable the identification of validated chemical probes. These biological efforts are coupled with structural determination and chemical optimization to reveal insights into the dynamic and flexible ligand binding properties of PXR, CAR, CYP3A4 and CYP3A5.
In so doing, we have developed two strategies to prevent PXR activation with a concrete structural activity relationship. We also established that PXR and CAR form an unexpected heterodimer, and we discovered the first CYP3A5-selective inhibitor and unraveled its structural basis. These achievements lay a solid foundation for designing new therapeutic strategies to overcome drug toxicity and resistance.
Selected publications
Contact us
Taosheng Chen, PhD, PMP
Chemical Biology & Therapeutics
MS 1000, Room E9054
St. Jude Children’s Research Hospital
Memphis, TN, 38105-3678 USA GET DIRECTIONS