St. Jude Family of Websites
Explore our cutting edge research, world-class patient care, career opportunities and more.
St. Jude Children's Research Hospital Home

- Fundraising
St. Jude Family of Websites
Explore our cutting edge research, world-class patient care, career opportunities and more.
St. Jude Children's Research Hospital Home
- Fundraising
If link downloads do not work, copy and paste into a new tab or window.
Performance is best in the Firefox browser.
CREST (Clipping Reveals Structure) is a new algorithm for detecting genomic structural variations at base-pair resolution using next-generation sequencing data. Please cite the following article:
Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, Holmfeldt L, Payne-Turner D, Fan X, Wei L, Zhao D, Obenauer JC, Naeve C, Mardis ER, Wilson RK, Downing JR and Zhang J. CREST maps somatic structural variation in cancer genomes with base-pair resolution (2011). Nature Methods.
The source code can be downloaded here and used according to the terms of the GNU General Public License (GPL), version 2 or later. Users will need to obtain the BLAT and CAP3 programs separately to use CREST; BLAT and CAP3 are free for academic use but require licensing fees for commercial use. The open source BioPerl and SAMtools libraries are also needed to use CREST.
Download CREST 1.0 Program. (540 KB)
hg18.fa (3.1 GB) and hg18.2bit (773 MB): These large files are needed to run CREST on the test data provided with its download.
- BLAT for academic use: http://www.soe.ucsc.edu/~kent
- BLAT commercial license: http://www.kentinformatics.com/
- CAP3 for academic use: http://seq.cs.iastate.edu/cap3.html
- CAP3 commercial license: Contact Robin Kolehmainen at Michigan Tech, rakolehm@mtu.edu or (906) 487-2228
- BioPerl: http://www.bioperl.org/
- SAMtools: http://sourceforge.net/projects/samtools/files/
FAQ
CREST 1.0.1 Release Notes:
Introduction: This is a bug fix release, which has fixes for 3 reported bugs.
Changes:
- Fixed bug in bin_search
- Fixed bug in identification of the chromosome name when it has | in it, now it only requires the chromosome name has no ":" and "-" in the name.
- Fixed bug that when SV happens on a chromosome not in bam file, the SV is considered as invalidate and the program will not exit.
Q: Error message:
MSG: Unable to sort hits: Can't call method "start" on an undefined value at /usr/lib/perl5/site_perl/5.8.8/Bio/Search/SearchUtils.pm line 508,
A: This error is due to the BioPerl version compatibility. To remove this error message, just comment out line 151 and 152 of SVExtTools.pm:
151# $result->sort_hits(sub {$Bio::Search::Result::ResultI::b -> matches('id') <=>
152# $Bio::Search::Result::ResultI::a ->matches('id')});
Q: Insertion is identified as DEL in CREST.
A: The definitions of DEL, INS, ITX, INV and CTX are stated on the paper. CREST defines those events only from a focal point of view and just considers the relationship before and after the break points of the fusion sequence. So the INS definition is more like tandem duplication.
Q: I only got a handful of SVs from CREST, is this normal?
A: For a normal sample, overall you will see over one thousand SVs compared to the reference genome. For somatic SVs, the number of SVs is related to the genome stability/complexity and can range from just a few to thousands from our experience. CREST overall does not report too many SVs as stated in the paper. However, low tumor cellularity (i.e. normal contamination in tumor) may affect the ability for detecting SVs as CREST requires at least 3 soft-clipping reads for each SV.
Q: CREST complains blat server is not accessible or gives segmentation fault, what’s the problem?
A: Please follow the steps from the README file in blat software package. After installing gfServer, use gfClient to check that the server is properly installed. Sometime you may need help from a system administrator to make sure the port you are using is open for access. Also, please give the full path to the 2bit file.
Q: Is this normal to see output from CREST like:
SV filter starting.... low complexity filter Type distance filter Germline sclip filter Loaded 201 letters in 1 sequences Searched 136 bases in 3 sequences Germline INDEL FILTER test Mapping quality filter ... FAILED
A: Yes, it’s normal. CREST will output each filter it is using and when any of the filters fails, the corresponding SV is considered false. When you see PASSED in the output, it means CREST identified a “true” SV.
Q: I noticed a disproportionate amount of INV/ITXs in the output, is it normal?
A: This may indicate an issue with library construction. We noticed this problem when analyzing some of our own PCGP samples. Interestingly, samples with excessive number of INV/ITX are likely to have uneven coverage across the genome, creating a feature that we termed “fractured genome”. We selected one such sample and re-prepared the library and did the whole-genome sequence. Both the uneven coverage and the excessive INV/ITX disappeared in the second experiment, indicating that the “fractured” genome was caused by artifacts. It is possible that DNA segments with INV/ITX were circulated and then cut in random positions before the sequencing adaptors were added. Those circulated segments will be identified as ITX/INV, but the distance between the break points should be small (relative to the insert size), and you should not expect a big portion of reads cross the break points show this feature.
The bottom line is that a large number of small INV/ITX is mostly likely to be an artifact caused by library construction.
Q: Error message:
Use of uninitialized value $sdna in substr at /usr/local/lib64/perl5/Bio/DB/Bam/AlignWrapper.pm line 243
A: Usually this problem is due to the inconsistency of the genome files used to do mapping and to do SV detection (for example, one is from hg18 and another is from hg19). Make sure the exact same file is used and the 2bit file is generated from this genome file.
Q: Error message:
Use of uninitialized value $seq in substr at /usr/local/lib64/perl5/Bio/DB/Bam/AlignWrapper.pm line 274
A: The error is due to the inconsistency between Bio::DB::Sam and the mapping tools (bwa etc) on how to deal with soft-clipping reads in the MD tag. bwa sometimes still give you mapping information (insertion, deletion, matches and mismatches) for cigar character ‘S’, while Bio::DB::Sam think you should not give any mapping information for ‘S’ and you should skip it. The solution is to just change the dna method in AlignWrapper.pm; more precisely add the following line:
return $self->{sam}->seq($self->seq_id,$self->start,$self->end);into line 253 of AlignWrapper.pm.
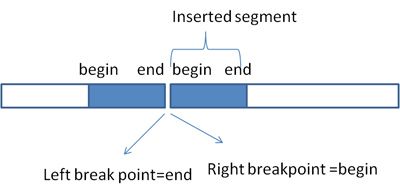
Q: I was puzzled by the coordinates presented in *.predSV.txt files. I found many insertion events as below:
chrX 55678965 + 13 chrX 52886736 + 0 INS 26 24 55 0 0.730555555555556 0.583333333333333 1 0 1 chrX 55678880 141 chrX 52886790 ACATACTCTTTTGTCTTTGTCTTTATGCCCGTGTTCATCCTCCTTTGTTCAGTCCAGCAAGGTCTGCAGCATTATAAAGTTCAAAGGCA TGGGAACCTAGAGCTGCCCCTTCTGTCTTTCTTTTAAGTAAGGTCCAAAGGT
The README of crest, the 2nd column represents left_pos and the 6th column means right_pos, but the coordinate of 2nd column is larger than that of 6th column, and the insertion spans (55678965-52886736)=2792229(bp), which I couldn’t understand. As I know, insertion always has the same coordinates for start and end position on the reference. Is there something I misunderstand? Does the crest not tell the start and end position of an SV event to usrs directly?
A: The type “INS” refers to a duplication event where the signature is generated by having the end of duplicated segment “abutted” to its head. This is why you will have a left breakpoint with the low genomic coordinate while the right breakpoint with a high genomic coordinate. This is illustrated below: